Сильный комбинированный иммунный дефицит представляет собой группу редких врожденных синдромов с небольшими иммунными ответами или без них. Это приводит к частым заражения бактериями, грибками и вирусами. Инфекции, которые являются незначительными для большинства людей, могут быть опасными для жизни у людей с синдромами иммунодефицита, связанными с комбинированным дефектом клеточного и гуморального иммунитета. Иммунная система включает в себя специализированные лейкоциты, которые работают вместе для борьбы с бактериями, грибами и вирусами. Эти лейкоциты включают Т-лимфоциты (Т-клетки), которые являются центральными медиаторами иммунного ответа, а также непосредственно атакуют вирусы, B-лимфоциты (В-клетки) продуцируют антитела, которые прикрепляются к инородным телам и отмечают их как фагоциты. NK-клетки также специализируются на борьбе с вирусами.
Пациенты с синдромами иммунодефицита, связанногое с комбинированным дефектом клеточного и гуморального иммунитета, имеют генетический дефект, который поражает Т-клетки и по меньшей мере один другой тип иммунной клетки. Существует несколько типов синдромов иммунодефицита, каждый из которых вызван другим генетическим (наследственным) дефектом.
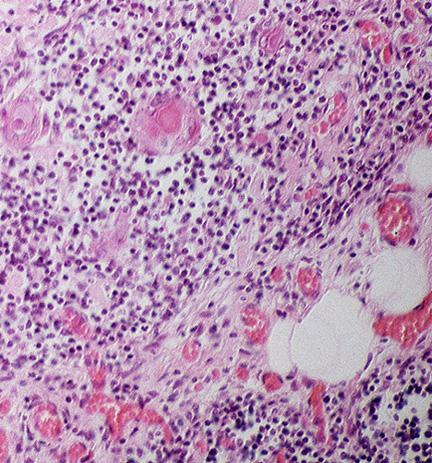
Этот пациент с аутосомно-рецессивным типом тяжелого комбинированного иммунодефицита умер от цитомегаловирусной пневмонии в возрасте 22 месяцев после предшествующих инфекций, включая рецидивный отит, пневмонию и оральную молочницу.
Сильный комбинированный иммунный дефицит, также называемый синдром Гландзмана и Риккера, представляет собой угрожающий жизни рецидивирующий синдром, диарею, дерматит. Это прототип первичных иммунодефицитных заболеваний и вызван рядом молекулярных дефектов, которые приводят к серьезному повреждению числа и функции Т-клеток, В-клеток и иногда NK-клеток. Без вмешательства синдром обычно приводит к тяжелой инфекции и смерти у детей в возрасте до 2 лет.
Тяжелый комбинированный иммунный дефицит является результатом мутаций в одном из более чем 15 известных генов. Эти молекулярные дефекты мешают развитию и функционированию лимфоцитов, блокируя дифференцировку и пролиферацию Т-клеток, а также в некоторых типах В-клеток и NK-клеток. Производство антител сильно нарушено, даже когда зрелые В-клетки присутствуют из-за отсутствия поддержки Т-клеток. NK-клетки, составляющие врожденного иммунитета, являются переменными. Последовательность и другие методы могут выявить фактические генетические дефекты у этих пациентов.
Патогенез тяжелого комбинированного иммунного дефицита можно разделить на следующие 5 механизмов, основанных на стадиях повреждения лимфопоэза:
- Дефектный сигнал лимфоцитов — ранний блок может появляться в пути дифференцировки Т-клеток. Наиболее распространенной формой синдрома, возникающей у 40-60% пациентов, является XL-форма, которая возникает из-за дефектов в общей цепи интерлейкина-рецептора. Этот молекулярный дефект приводит к отсутствию зрелости Т-клеток и NK-клеток.
- Апоптоз из-за накопления токсичных метаболитов — аномальный пуриновый обмен может быть вовлечен в патогенез синдрома Гландзмана и Рикера. Это приводит к накоплению промежуточных продуктов (например, аденозиндифосфата, гуанозинтрифосфата), что приводит к токсичности лимфоцитов, особенно у незрелых тимусных лимфоцитов.
- Дефектная сигнализация клеток — CD45, тирозинфосфатаза, обнаруженная в клеточных мембранах гемопоэтических клеток, необходима для регулирования передачи сигналов поверхности клетки в В-клетках и Т-клетках.
- Может произойти неправильная перегруппировка генов TCR и Ig — аномальная перегруппировка генов TCR и Ig. Как созревание В-клеток, так и созревание Т-клеток связаны с процессом рекомбинации, в котором собираются различные комбинации генов VDJ для создания уникальных и специфических рецепторов антигена. Несколько рекомбинаций играют важную роль в этом процессе.
- Thymic Dysgenesis — приводит к отсутствию Т-клеток
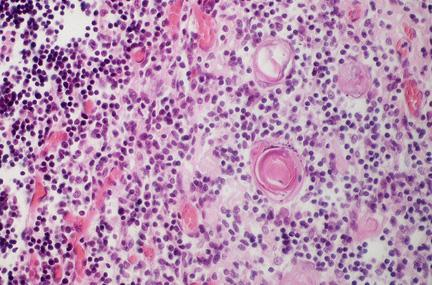
Гистологически, тимус при тяжелом комбинированном иммунодефиците, как правило, не содержит Hassall corpuscles и истощается лимфоцитами. На этой фотографии Hassall corpuscles справа от центра.
В классическом синдроме Гландзмана и синдроме Рикнера, связанного с синдромами иммунодефицита, связанными с комбинированным дефектом клеточного и гуморального иммунитета, тимус является гипопластичным. Кожа и кишечник могут проявлять инфильтрацию гистиоцитами, эозинофилами или активированными дисфункциональными Т-клетками. Эпидермис может иметь очаговые области гиперкератоза с паракератозом или нерегулярным акантозом с спонгиозом и экзоцитозом. Папиллярная дерма имеет отек и диффузные периваскулярные инфильтраты с некоторыми эозинофилами.
Селезенка и периферические лимфатические узлы обычно атрофичны, но иногда они могут быть гиперпластическими, с гистиоцитами и эозинофилами. Селезенка истощается лимфоцитами. Хотя биопсия лимфатических узлов не требуется для диагностики, результаты могут указывать на отсутствие Т- и В-клеток и отсутствие центров зародышей. Миндалины и аденоиды недоразвиты или отсутствуют.